Fibroblast Growth Factor Receptors (FGFR) and carcinogenesis
At the recent International Society of Gastroenterological Carcinogenesis (ISGC) meeting hosted by MD Anderson Cancer Centre that I attended in Houston, one of the topics mentioned the potential role of Fibroblast Growth Factor Receptors (FGFR) in carcinogenesis. I thought this was a great opportunity to research the area further.
A recent review of the role of FGFR in carcinogenesis fortuitiously appeared in Molecular Cancer Research:
“The fibroblast growth factor receptors (FGFR) play essential roles both during development and in the adult. Upon ligand binding, FGFRs induce intracellular signaling networks that tightly regulate key biological processes, such as cell proliferation, survival, migration, and differentiation. Deregulation of FGFR signaling can thus alter tissue homeostasis and has been associated with several developmental syndromes as well as with many types of cancer.
In human cancer, FGFRs have been found to be deregulated by multiple mechanisms, including aberrant expression, mutations, chromosomal rearrangements, and amplifications.”
They also went on to define what carcinogenesis is based on what we learned from Hanahan and Weinberg (2000) in their classic paper, The hallmark of Cancer:
“Carcinogenesis is a multistep process during which normal cells are transformed into cancer cells by accumulating several genetic changes and acquiring several common features that promote the malignant phenotype, often referred to as the hallmarks of cancer.
The six classic hallmarks of cancer include self-sufficiency in growth signals, insensitivity to antigrowth signals, limitless replication, evasion of apoptosis, sustained angiogenesis, and the ability to invade tissue and form metastasis.”
Bearing in mind that Hanrahan and Weinberg’s paper was published over a decade ago, there were some interesting observations that hold true:
“It is increasingly apparent that the growth deregulation within a tumor can only be explained once we understand the contributions of the ancillary cells present in a tumor—the apparently normal bystanders such as fibroblasts and endothelial cells—which must play key roles in driving tumor cell proliferation.”
Their article is now open access, so you can see how they described the role of these cells with cancer cells.
Going back to Haugsten et al., (2010) review, we learn new developments in carcinogenesis that have taken place over the last decade relating to FGFR, which has received much less attention than other receptors such as VEGF, PDGF, EGFR and IGF-1R as a potential target. The FGFR family consists of four genes encoding the tyrosine kinase receptors (FGFR1 to FGFR4). Downstream, the pathway becomes quite complex, so it will be interesting to see which factors emerge as escape routes through cross-talk and feedback loops.

The potential role of FGFR in cancer carcinogenesis is summarised in the table below:
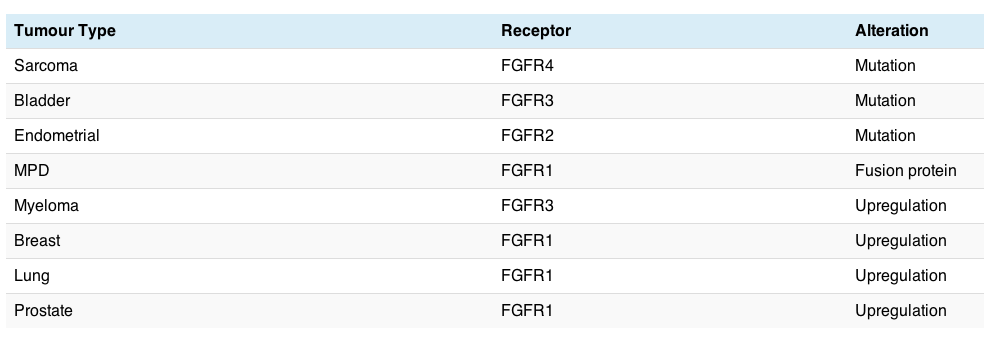
There are a variety of different cancers potentially affected, but FGFR is not always overexpressed where it is amplified and may not always be contained in the amplification. Sometimes overexpression merely indicates a poorer prognosis or the development of resistance.
The chances of it being a drugable target and therefore more likely to have a meaningful clinical impact is probably greater where there are mutations or fusion proteins, since these often (but not always) have aberrant activity associated with them. The challenge is therefore figuring where it might be a passenger or a driver gene.
Turner and Grosse (2010) also reviewed the FGFR pathway as a new area of research:
“Although FGF signalling can drive tumorigenesis, in different contexts FGF signalling can mediate tumour protective functions. The identification of the mechanisms that underlie these differential effects will be important to understand how FGF signalling can be most appropriately therapeutically targeted.”
Obviously, we won’t know the answer until clinical trials with the small molecule inhibitors and monoclonal antibodies are completed, but it certainly looks to be a worthwhile area of exploration. Hynes and Dey (2010) discussed the potential role in breast cancer in more detail, noting the findings of Roidl et al (2009):
“In breast cancer cell lines, it has been reported that increased levels of FGFR4 are found in cells resistant to chemotherapeutics.”
This suggests a combination strategy in those cases may be worthwhile.
For those who missed it, I also posted about the potential role of FGFR1 in lung cancer last month, which also included some of the inhibitors emerging in this class.
References:
![]() Haugsten, E., Wiedlocha, A., Olsnes, S., & Wesche, J. (2010). Roles of Fibroblast Growth Factor Receptors in Carcinogenesis Molecular Cancer Research, 8 (11), 1439-1452 DOI: 10.1158/1541-7786.MCR-10-0168
Haugsten, E., Wiedlocha, A., Olsnes, S., & Wesche, J. (2010). Roles of Fibroblast Growth Factor Receptors in Carcinogenesis Molecular Cancer Research, 8 (11), 1439-1452 DOI: 10.1158/1541-7786.MCR-10-0168
Hanahan, D., & Weinberg, R. (2000). The Hallmarks of Cancer Cell, 100 (1), 57-70 DOI: 10.1016/S0092-8674(00)81683-9
Turner, N., & Grose, R. (2010). Fibroblast growth factor signalling: from development to cancer Nature Reviews Cancer, 10 (2), 116-129 DOI: 10.1038/nrc2780
Hynes, N., & Dey, J. (2010). Potential for Targeting the Fibroblast Growth Factor Receptors in Breast Cancer Cancer Research, 70 (13), 5199-5202 DOI: 10.1158/0008-5472.CAN-10-0918
Roidl A, Berger HJ, Kumar S, Bange J, Knyazev P, & Ullrich A (2009). Resistance to chemotherapy is associated with fibroblast growth factor receptor 4 up-regulation. Clinical cancer research : an official journal of the American Association for Cancer Research, 15 (6), 2058-66 PMID: 19240166