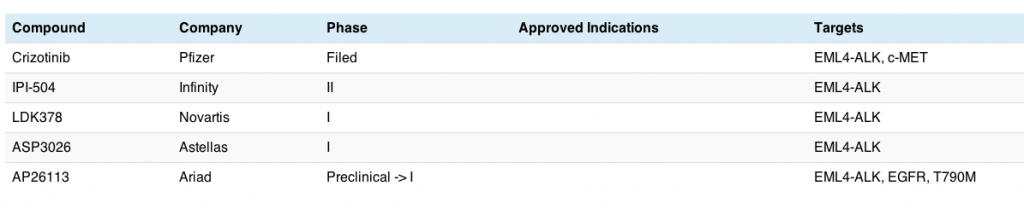
Yesterday we discussed ELM4-ALK translocations associated with adenocarcinomas in non-small cell lung cancer (NSCLC). Before ALK was identified, the main molecular target in this disease was EGFR mutations, which have been shown to be associated with improved responses when EGFR inhibitors such as erlotinib are given.
EGFR mutations
Although erlotinib is the EGFR inhibitor of choice in the US for patients with lung cancer who have EGFR mutations, unfortunately approx. 50% of them with development resistance as a new mutation evolves, known as T790M. In addition, patients with EGFR mutant disease generally don’t respond well to erlotinib.
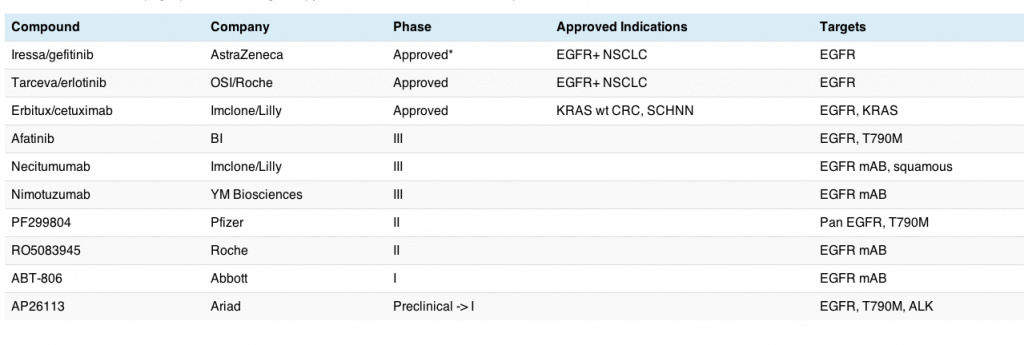
There are a number of EGFR inhibitors now in development for lung cancer, specially adenocarcinomas, and this market is rapidly getting fairly crowded, as the table shows:
Note: *Although gefitinib was approved, there is very little use of this agent in the US since the FDA modified the PI to include mention of no overall survival benefit.
This table does not include all EGFR inhibitors in clinical development, since those focused on HER2+ breast and gastric cancers (trastuzumab, lapatinib, pertuzumab) and KRAS wt colorectal cancer (cetuximab and panitumumab) were excluded from this analysis. There are, no doubt, other EGFR inhibitors also being evaluated in lung cancer, but these are the main ones I’m following – it’s for illustrative purposes only!
What we can see here is that the EGFR market in lung cancer is getting pretty competitive in terms of clinical trials and is a much more mature market than the ALK one discussed yesterday, although only erlotinib is still currently available in the US for treatment of EGFR+ mutations.
Beyond that, there are at least three inhibitors in phase III trials alone:
- Afatinib, which reported rather indifferent data in the LUX-LUNG1 trial at ESMO last September, with an improvement in response rate over placebo, but no advantage in overall survival, the primary endpoint of the trial.
- Necitimumab, a monoclonal antibody being tested in squamous histology in combination with gemcitabine and cisplatin.
- Nimotuzumab, another monoclonal antibody to EGFR, which is in phase III for oesophageal and H&N cancers, but currently enrolling in several phase II lung cancer trials at the moment.
Aside from the sheer awkwardness of trying to say either of the last two generic names and resorting, like many people to calling them Nessie and Nemo, I also have trouble remembering which one is which or my eyes glaze over! The tongue twisters that are cropping up for a multitude of targeted therapies is more challenging than ever. On top of that is the dreadful tendency of late to choose ugly brand names for cancer drugs that seem to have been purchased as a job lot from Eastern Europe brand agencies.
T790M confers resistance to erlotinib
We have known for a while that EGFR inhibitors do not work in all patients:
- They are effective in wild type but not mutant EGFR
- Some patients on EGFR therapy develop resistance due to a new mutation, T790M, appearing (see Hammerman et al., 2009).
There has been little progress in either of these two areas of late, although a multitude of clinical trials are now ongoing and we are awaiting data readouts.

Dr Jack West, Swedish Cancer Institute
Dr Jack West from the Swedish Cancer Institute in Seattle blogged about his enthusiasm for the irreversible EGFR inhibitor, PF299804, in lung cancer last summer, based on the two small studies (one American and one Asian) that were reported at the time. Another small Australian study on EGFR mutations and PF299804 was also presented at ASCO last year.
The Asian study looked promising enough at the time for Park et al., (2010) to conclude that the data from the Korean patients with KRAS wild-type NSCLC (adenocarcinoma histology) who were refractory to platinum-based chemotherapy and erlotinib (E) or gefitinib (G) that:
“Preliminary data consistent with Western trials show that in heavily treated Asian pts with NSCLC after E or G failure, PF299 is well tolerated and has antitumor activity without adversely impacting pts’ HRQOL. These results support the ongoing global P3 trial in pts with refractory NSCLC.”
KRAS mutations in NSCLC
I distinctly remember at ASCO in 2010 one of the three groups stated that a phase III trial (BR26) would be forthcoming in KRAS wt patients. This trial is is now enrolling patients in the refractory setting after failure of erlotinib in both KRAS mutant and wild type patients, so we are unlikely to get a readout of this study before the end of 2012 at the earliest.

Dr Nathan Pennell, Taussig Cancer Center
What was also interesting was another more recent post on GRACE by one of the faculty, Dr Nathan Pennell, discussing the combination of afatinib and cetuximab in EGFR+ lung cancer.
Like Dr Pennell, I would not have been enthusiastic about this potential combination after seeing the LUX-LUNG data presented by Dr Vincent Miller at ESMO last September, and knowing that cetuximab has shown weak activity in lung cancer at best, both alone and in combination with erlotinib.
However, as Dr Pennell described, the results with the afatinib plus cetuximab combination were much better than expected in erlotinib refractory lung cancer patients:
“The confirmed overall response rate in the first 45 evaluable NSCLC patients was 40%, with the response rate in the T790M patients being 50% unconfirmed (unconfirmed means they had not yet done a second set of scans to see if the tumor was still responding over 2 time points).”
Understandably, he was quietly excited by the outcome:
“This is the first time anyone has described activity for any targeted drug in the T790M EGFR population, and this provides the first glimmer of hope for the many patients out there who face the reality of knowing that their tumors are still dependent on EGFR mutations but for whom Tarceva simply doesn’t work anymore.”
This is most interesting, because while afatinib is a TKI that works similarly to erlotinib and gefitinib inside the cell on the EGFR ligand, cetuximab is a monoclonal antibody that works on the outside of the ligand, suggesting that targeting the ligand upstream and downstream may well be necessary to ensure complete EGFR inhibition in these patients ie EGFR, KRAS and T790M, we don’t know for sure, but now doubt a paper will be published soon explaining the scientific phenomenom.
The result is certainly a big surprise given the previous disappointing data for cetuximab combined with erlotinib (no responses out of 19 patients evaluated), so I’m not sure why afatinib is different given it is also a small molecule TKI. Further large scale trials will needed to confirm these promising results. A phase I trial with cetuximab and afatinib in NSCLC is currently enrolling patients.
For those interested, you can check out the simply stunning waterfall plot that Dr Pennell found for the combination of afatinib and cetuximab, with both EGFR+ and EGFR- patients responding. I confess I nearly fell off my chair when I looked – the data is not what was expected at all!
Meanwhile, Pfizer also appear to be looking at PF299804 in HER2+ gastric cancer, which is logical given the activity previously demonstrated by trastuzumab. There are over 20 studies with this pan-EGFR agent. Although I’m sure I read somewhere that PF299804 specifically targets the T790M mutations that develop with erlotinib, it will be interesting to see what happens with wild-type and mutant subsets, since we know that erlotinib and gefitinib only work in wild type not mutant EGFR. Another clinical trial is also enrolling patients with Pfizer’s pan-EGFR (PF299804) and MET inhibitor (crizotinib) in NSCLC. More on c-MET inhibitors will follow in another post.
All that said, the rationale for targeting T790M mutations in erlotinib-refractory disease is fairly compelling in theory, but the trials will not be easy as there are several challenges that must be overcome:
- Design and implementation – patient and inhibitor selection will be crucial
- High risk, especially if the compound is a weak inhibitor
- Heavily pre-treated refractory patients tend to be sick with a higher tumour burden that is more resistant to therapy
- It is a small, difficult to treat subset
- Finding patients with good performance status who meet all the eligibility criteria is never easy in small refractory subset populations
- Would combinations be superior to single agent therapy or should sequencing be investigated?
- An upfront trial as a head to head with erlotinib would clearly be easier to execute, but also high risk if the agent is too similar
The more you look at the conundrum, the more one realises that addressing it is not as easy as it first looks. Some basic research looking at sequencing versus combinations in cells lines and xenografts with different agents may be a useful starting point.
That said, the news last Friday that Ariad are advancing their dual EGFR-ALK inhibitor AP26113 into the clinic later this year is good news for patients with lung cancer, because more available options is always better than fewer. It will be interesting to see how the drug fares in either ALK-refractory patients (refractory to crizotinib) or in EGFR mutations (refractory to erlotinib) as potential fast track to market strategies.
Conclusions
Overall, the landscape in lung cancer is rapidly changing as new molecular targets are being identified, along with new targeted therapies that specifically inhibit the driving cancerous activity.
EGFR mutations were the first major breakthrough in targeted therapies for lung cancer, but with the discovery of ALK translocations, T790M mutations, MET and FGFR1 ampliflication, and DDR2 mutations, we now have exciting opportunities to potentially match therapies to patients in across a range of different non-small cell lung cancer subsets in a new era of molecularly targeted and personalised therapy.
Beyond erlotinib, there is a clear high unmet medical need for new targeted therapies that specifically inhibit either the T790M mutation associated with resistance to initial therapy or will work in KRAS mutant EGFR lung cancers. The afatinib + cetuximab combination in erlotinib refractory NSCLC looks very promising and I’m very much looking forward to watching all the new developments evolve in this area.
Disclosure: I am an unpaid member/volunteer of the GRACE Board.
References:
 Hammerman PS, Jänne PA, & Johnson BE (2009). Resistance to Epidermal Growth Factor Receptor Tyrosine Kinase Inhibitors in Non-Small Cell Lung Cancer. Clinical Cancer Research, 15 (24), 7502-7509 PMID: 20008850
Hammerman PS, Jänne PA, & Johnson BE (2009). Resistance to Epidermal Growth Factor Receptor Tyrosine Kinase Inhibitors in Non-Small Cell Lung Cancer. Clinical Cancer Research, 15 (24), 7502-7509 PMID: 20008850