Update on RAF resistance in metastatic melanoma
“RAF inhibitors (vemurafenib and dabrafenib) have profound clinical activity in patients with BRAF-mutant melanoma, but their therapeutic effects are limited by the emergence of drug resistance.”
Solit and Rosen (2014)
For today’s post on Science Fridays, I wanted to take a look at an overview paper, published in Cancer Discovery, from two researchers in the metastatic melanoma field who have been looking at multiple mechanisms of resistance. It’s an important topic because while we have seen incremental improvements in outcomes for this disease, the 5-year survival rate is still rather poor with only 10–20% of metastatic patients still alive by then. This is not to disparage the efforts of scientists, clinicians or companies working in this space, far from it, but there is is clearly a need for new therapies, strategies and combinations, given the high unmet medical need that exists.
We still have a long way to go in moving the survival needle dramatically.
It wasn’t until I searched for related blog posts to link to this one that I realised how much we have already covered on this topic! Regular readers will recall discussions here on PSB on various combinations such as:
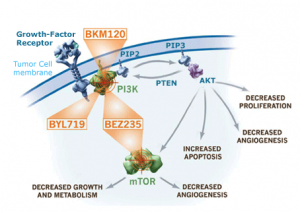
- RAF + MEK inhibitors (downstream resistance)
- RAF + PI3K-AKT-mTOR inhibitors (cross resistance)
- RAF + CTLA–4 checkpoint inhibitors (anti-tumour immunity)
to name a few examples.
We have seen that adding a MEK inhibitor to dabrafenib e.g. trametinib can overcome resistance temporarily and add a few extra months before the resistance sets in again. Similarly for PI3K inhibitors tested to date. Adding ipilimumab, an anti-CTLA–4 checkpoint inhibitor held much promise, but the combination was abandoned with the emergence of unexpected liver toxicity.
Results thus far suggest that something else is acting as an escape route, thereby enabling the tumour to continue driving oncogenic addiction to BRAF.
The $64K questions are what is happening and what can we do about it?
We also need to remember that clinical research advances piecemeal based on evidence from preclinical reseach, so we see the logical evolution of BRAF monotherapy -> combos with downstream (MEK) or upstream (NRAS) targets in same pathway -> combos with diagonal (PI3K) pathways etc.
What Solit and Rosen have done is put a nice summary together of the state of play in this disease and the paper (see References below) is well worth reading.
Their main assertion is interesting, namely:
“The common feature of each of these mechanisms of resistance is that they result in activation of ERK signaling that is insensitive to the RAF inhibitor. Thus, RAF inhibitor resistance is often associated with maintenance of activation of the oncogene-driven pathway.”
Two recent papers are cited in support of this theory from Shi et al., (2014) and Allen et al., (2014) – see References below for additional background reading. Both studies used patient samples to look at clonal evolution and the genetic landscape in advanced melanoma. It’s actually quite amazing what unbiased exome sequencing can uncover at the molecular level, not least are the development of new mutations and other functional alterations.
The Shi et al., (2014) study was briefly summarised by Solit and Rosen:
“Multiple biopsies were obtained at different times or from disparate locations from several patients, and more than a single lesion in the ERK pathway was identifi ed in multiple patients typically within
different tumor biopsies.”
They went to note:
“A detailed phylogenetic analysis of multiple progressive lesions from a subset of these patients suggested branching evolution of tumors in which the development of genetic diversity was not linearly associated with time.”
Previously, a case report found distinct mechanisms of BRAF inhibitor resistance were present in two different progressing lesions from a single patient, so the work of Shi et al., (2014) is consistent with this finding. It blows my mind that different lesions in the same patient might behave completely differently though – imagine trying to devise an appropriate and effective clinical strategy in these cases?!
Allen et al’s (2014) work also involved whole exome sequencing (WES) from patient samples:
“WES was performed on paired pretreatment and progression samples collected from 45 patients, of whom 14 developed resistance soon after initiation of therapy (within 12 weeks). They also detected several resistance mechanisms that had been previously identified to confer RAF inhibitor resistance, including mutations in NRAS , MAP2K1, and NF1 and BRAF amplification.”
A third important study in this area from Wagle et al., (2014) adds to the weight of evidence with new mutations developing. Solit and Rosen continued the story:
“Consistent with the preclinical studies highlighted above demonstrating that MEK1 and MEK2
mutations can confer RAF and MEK inhibitor resistance, a MEK2 Q60P mutation was identifi ed in 1 of 5 patients studied. Of greater surprise to the investigators, one patient had a BRAF splice variant lacking exons 2–10 and a second patient had BRAF amplification.”
By now, you can see the sheer variety of changes and adaptations taking place in different studies around the world in some of the top melanoma labs. What do they have in common though?
“One hypothesis to explain this result is that increased abundance of the oncogenic driver (in this case BRAF) in response to prolonged drug treatment results in increased flux through the ERK pathway and restoration of ERK activity above the threshold required for inhibition of cell proliferation.”
The next challenge is to figure how we can approach better therapeutic index and shutting down of the pathways?
“The results suggest that the early adaptive response of BRAF -mutant cells to ERK pathway inhibition may promote the selection of resistant clones that harbor additional genomic events that
confer higher levels of RAF inhibitor resistance. The data also support combinatorial approaches that attenuate the adaptive response, including the addition of a PI3K or AKT inhibitor to the RAF and MEK (or ERK) inhibitor combination.”
The problem with this approach though, is that the neither the BRAF nor PI3K inhibitors have been able to reach or go beyond the single agent dosing schedules:
“As previous attempts to combine MAPK and PI3K pathway inhibitors have been limited by overlapping toxicities, upfront testing of intermittent treatment schedules should be considered.”
This is the also approach that Das Thakur suggested in her work presented at AACR last year, and subsequently published in Nature, to delay the development of resistance to vemurafenib.
I do think this one area where we may well see new trials evolve in advanced melanoma, so we will have to wait for new data before we can see if the strategy is successful at delaying the emergence of resistant clones. It is good to see the evolution of solid preclinical and translational evidence from patient biopsies helping to inform future clinical trial strategies.
In the meantime, the next major milestone I’m waiting for is on Roche/Genentech’s MEK inhibitor, cobimetinib (GDC–0973), which is due to report combination data with vemurafenib (continuous dosing) later this year. It will be interesting to see if this inhibits MEK more completely than trametinib and whether the combination has a better initial outcome than dabrafenib plus trametinib, which added about two to three months of extra survival over dabrafenib alone.
References:
![]() Solit DB, & Rosen N (2014). Towards a Unified Model of RAF Inhibitor Resistance. Cancer discovery, 4 (1), 27–30 PMID: 24402945
Solit DB, & Rosen N (2014). Towards a Unified Model of RAF Inhibitor Resistance. Cancer discovery, 4 (1), 27–30 PMID: 24402945
Shi H, Hugo W, Kong X, Hong A, Koya RC, Moriceau G, Chodon T, Guo R, Johnson DB, Dahlman KB, Kelley MC, Kefford RF, Chmielowski B, Glaspy JA, Sosman JA, van Baren N, Long GV, Ribas A, & Lo RS (2014). Acquired Resistance and Clonal Evolution in Melanoma during BRAF Inhibitor Therapy. Cancer discovery, 4 (1), 80–93 PMID: 24265155
Van Allen EM, Wagle N, Sucker A, Treacy DJ, Johannessen CM, Goetz EM, Place CS, Taylor-Weiner A, Whittaker S, Kryukov GV, Hodis E, Rosenberg M, McKenna A, Cibulskis K, Farlow D, Zimmer L, Hillen U, Gutzmer R, Goldinger SM, Ugurel S, Gogas HJ, Egberts F, Berking C, Trefzer U, Loquai C, Weide B, Hassel JC, Gabriel SB, Carter SL, Getz G, Garraway LA, Schadendorf D, & Dermatologic Cooperative Oncology Group of Germany (DeCOG) (2014). The Genetic Landscape of Clinical Resistance to RAF Inhibition in Metastatic Melanoma. Cancer discovery, 4 (1), 94–109 PMID: 24265153
Wagle N, Van Allen EM, Treacy DJ, Frederick DT, Cooper ZA, Taylor-Weiner A, Rosenberg M, Goetz EM, Sullivan RJ, Farlow DN, Friedrich DC, Anderka K, Perrin D, Johannessen CM, McKenna A, Cibulskis K, Kryukov G, Hodis E, Lawrence DP, Fisher S, Getz G, Gabriel SB, Carter SL, Flaherty KT, Wargo JA, & Garraway LA (2014). MAP Kinase Pathway Alterations in BRAF-Mutant Melanoma Patients with Acquired Resistance to Combined RAF/MEK Inhibition. Cancer discovery, 4 (1), 61–8 PMID: 24265154
Das Thakur M, Salangsang F, Landman AS, Sellers WR, Pryer NK, Levesque MP, Dummer R, McMahon M, & Stuart DD (2013). Modelling vemurafenib resistance in melanoma reveals a strategy to forestall drug resistance. Nature, 494 (7436), 251–5 PMID: 23302800