What’s hot at ECCO 2013 – Preview of late breakers and PI3K abstracts
Next I’ll be off to the European Cancer Congress (ECC) in Amsterdam. This meeting alternates each year between ECCO and ESMO hosting the event at a different European city.
The last couple of years have seen some nice data that missed the ASCO deadline, other years can bring an update of the already familiar ASCO data. I suspect that this year will be one of those events, with updated PD-1 and PD-L1 data.
If you missed my colleague Pieter Droppert’s ECCO highlights yesterday, you can catch them here, including details of the iPad app and abstracts.
In addition, there were other abstracts of interest that caught my eye, including some solid late breakers:
1. T-DM1 for HER2-positive metastatic breast cancer (MBC): Primary results from TH3RESA, a phase 3 study of T-DM1 vs treatment of physician’s choice. H. Wildiers (Belgium) et al.
The study looks at advanced disease in patients who had received at least two prior regimens. This analysis looks like an interim one, given the full study timeframe is over 3.5 years. I’m particularly curious what the physician choices were to compete with Kadcyla and what the 1 year survival curves look like. It’s a wee bit early to hope that they might separate already, certainly I hope they do!
2. Evaluation of everolimus (EVE) in HER2+ advanced breast cancer (BC) with activated PI3K/mTOR pathway: Exploratory biomarker observations from the BOLERO-3 trial. G. Jerusalem (Belgium) et al.
Originally, I thought this had been presented at ASCO, but the biomarker abstract I found actually referred to BOLERO-2, where they noted that “efficacy was greater in patients with low PI3K expression”, which is an odd finding. The BOLERO-3 data from ASCO presented the initial phase III data for the combination of trastuzumab, vinorelbine and everolimus vs trastuzumab and vinorelbine alone in trastuzumab resistant HER2+ advanced breast cancer. This should be an interesting presentation worth attending.
3. FLT1 gene variation as a major determinant of recurrence in stage I-III non-small cell lung cancer. F. Innocenti (USA) et al.
Many of us familiar with FLT3 in leukemia, but FLT1 is an interesting concept with very little data (or drugs) out there. I will be curious to see if this is a druggable target and where this approach might lead.
4. Pooled analysis of long-term survival data from phase II and phase III trials of ipilimumab in metastatic or locally advanced, unresectable melanoma. F. Hodi (USA) et al.
If you look at five year survival curves for advanced melanoma in the literature, it’s historically around 20% or so when patients have received IL-2, which is where I would expect ipilimumab to be. Trials with DTIC have shown a much lower rate, at around 8-9%. Not every patient is suitable for IL-2 though, so we may be seeing similar survival rates irrespective of the immunotherapy given, but with very different safety profiles.
One of my favourite cancer pathways is PI3K-AKT-mTOR. It’s dysregulated in some 80% of cancers yet we haven’t really seen a major breakthrough with these agents in solid tumours outside of the stunning Afinitor data in relapsed ER+ metastatic breast cancer from the BOLERO-2 study presented at ECCO in Stockholm two years ago.
There are many different permutations out there from single to dual inhibitors and also specific isoforms of alpha, beta, delta and gamma.
One of the challenges with targeting PI3K is that it activates feedback loops. Thus inhibiting PI3K in advanced prostate cancer activates the androgen receptor, while single agent use in advanced breast cancer can lead to activation of HER3. In addition, there have been mixed results with biomarkers and specific mutations/tumour suppressors to date such as PIK3CA and PTEN. This increases the complexity tremendously and therefore speaks to more careful trial selection based on inclusion criteria and also logical combinations to try and shut down the compensatory pathway.
I was therefore pleased to see a few trials reporting early phase I/II data in this vein:
P017: Evaluation of tolerability and anti-tumor activity of GDC-0032, a PI3K inhibitor with enhanced activity against PIK3CA mutant tumors, administered to patients with advanced solid tumors.
D. Juric, J.R. Infante, I.E. Krop, C. Kurkjian, M.R. Patel, R.A. Graham, T.R. Wilson, J.Y. Hsu, J. Baselga, D.D. Von Hoff
According to the abstract:
“GDC-0032 is an orally bioavailable, potent, and selective inhibitor of Class I PI3K alpha, delta, and gamma isoforms, with 30-fold less inhibition of the PI3K beta isoform relative to the PI3K alpha isoform.”
Several confirmed partial responses have been reported and further trials will continue:
“GDC-0032 is a next-generation PI3K inhibitor with promising anti-tumor activity observed in patients with PIK3CA mutant tumors. GDC-0032 is being investigated in combination with endocrine therapies such as letrozole and fulvestrant for patients with hormone receptor-positive breast cancer.”
P079: Hyperglycemia in patients treated with the pan-PI3K inhibitor buparlisib (BKM120): characterization, management, and assessment for pharmacodynamics
A. Azaro, J. Rodon, J.F. Vansteenkiste, Y. Ando, T. Doi, D. Mills, C. Sarr, E. Di Tomaso, C. Massacesi, R.W. Naumann
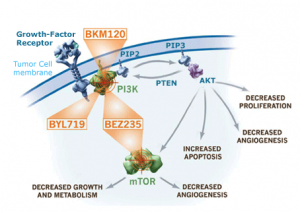
Source: Novartis
BKM120 is also an oral pan PI3K inhibitor that does not target mTOR. Aside from the activation feedback loop effects mentioned earlier, PI3K plays a key role in glucose homeostasis. A number of earlier trials with different PI3K and mTOR inhibitors have reported hyperglycemia as a class effect although they have varied in the degree to which the event occurred.
This study highlights the importance of a potential pharmacodynamic marker (C-peptide) in assessing the insulin response and I’m looking forward to seeing more detail in the poster.
P061: Factors predisposing to development of hyperglycaemia in phase 1 studies involving PI3K, mTOR, AKT and mTORC1 and mTORC2 inhibitors
M. Wong, K.H. Khan, K. Rihawi, S. Bodla, B. Amin, K. Shah, D. Morganstein, S.B. Kaye, U. Banerji, L.R. Molife
Related to the topic of hyperglycemia, the Royal Marsden mined their database for PI3K-mTOR trials and looked at factors that might influence the presence of the glucose spike in order to essentially try and predict which patients were more at risk and improve management. While most patients did not require intervention, but in those that did, metformin and insulin were usually preferred. Interestingly, the main factor emerging in this retrospective study was a prior history of diabetes, which is not totally unsurprising. It will be useful to see if these results can be validated in prospective future trials.
P227: Anti-tumour efficacy of the PI3K inhibitor GDC0941, the dual PI3K/mTOR inhibitor GDC0980 and the MEK inhibitor GDC0973 as single agents and in combination in endometrial carcinomas
O. Aslan, A.M. Farrelly, B. Stordal, B.T. Hennessy
Much has be written about the potential for a PI3K and MEK combination in different tumour types, but so far they haven’t proven to be the home run many of us hoped for.
This preclinical paper looks at cell lines to explore potential targets and synergies in endometrial cancer (EC). They concluded,
“Our data suggest that the mutational status of PIK3CA, PTEN and KRAS can be used as biomarkers to select patients for PI3K and RAS/RAF-targeted therapies. Further, the combinations of the PI3K inhibitors GDC0941 and GDC0980 with the MEK inhibitor GDC0973 are promising approaches for the treatment of patients with PIK3CA, PTEN and KRAS-mutated EC.”
Translating data from simple cell lines to complex human bodies does not always predict response given the variable responses seen from patients with mutations and tumour suppressors in clinical trials. I think it will take a while to tease out what defines and predicts a response in each tumour type much in the same way we saw different effects in advanced melanoma when targeting BRAF with sorafenib versus BRAF V600E with vemurafenib or dabrafenib. The devil is in the details.
And finally, an oral presentation with a very different focus in the PI3K related field that I’m really looking forward to hearing:
#1859 PI3KCA mutations and correlation with pCR in the NeoALTTO trial (BIG 01-06)
J. Baselga, I. Majewski, P.G. Nuciforo, H. Eidtmann, E. Holmes, C. Sotiriou, D. Fumagalli, M.C. Diaz Delgado, M. Piccart-Gebhart, R. Bernards
The authors evaluated:
“The influence of PI3K pathway mutations (PIK3CA, KRAS, BRAF, AKT1) on sensitivity to trastuzumab (T), lapatinib (L), or both agents (L+T) in combination in early-stage HER2-positive breast cancer patients enrolled the neoALTTO trial.”
The goal here is to see the presence of any of the mutations were more likely to lead to resistance and enable better selection of therapy for patients. I will update on this study after the presentation.
More detailed posts and synopses will continue from the meeting itself on Biotech Strategy Blog, where we’ll be sharing our insights and analysis daily.


