New solutions for resistance to therapies in lung cancer?
One of the things that is both frustrating and fascinating is the development of resistance to therapies in cancer treatment. By this, I mean clearly it’s not something we want to see from a patient or physician perspective and if possible, to delay it as long as feasible. On the other hand, the mechanics behind the biology of drug resistance is a fertile field for curious scientists.
I never fail to feel a sense of awe when a group cracks open new mechanisms that improve our understanding of cancer. It is, after all, a highly complex and fickle topic. I’ve often wondered why is it that some patients see resistance set in early and others do not? Why does resistance occur, period?
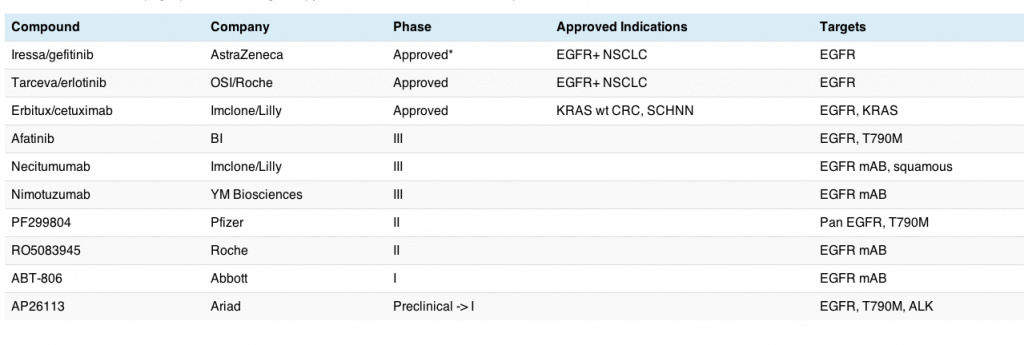
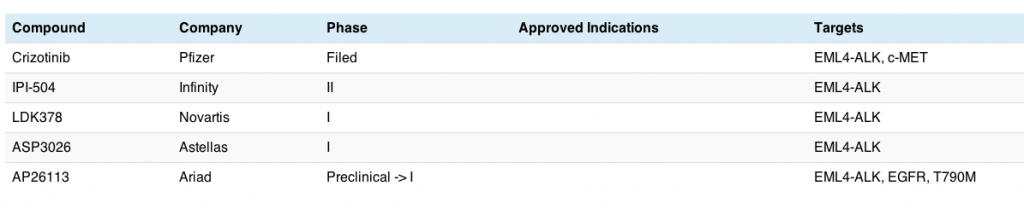
This morning my interest was piqued by a new paper published this month in Science Translational Medicine from William Pao’s group at Vanderbilt. They looked at the conundrum around EGFR inhibitors such as erlotinib, gefitinib and afatinib in non-small cell lung cancer (NSCLC) because patients treated with these drugs eventually develop acquired resistance to therapy and the cancer unfortunately starts growing again. The big question are why and what?
“The most common mechanism of resistance is a second site mutation within exon 20 of EGFR (T790M), observed in ~50% of cases. This change leads to altered binding of the drug within the ATP pocket.”
In this elegant research, they looked at the behaviour in cell lines before and after the cells acquire resistance to targeted therapy:
“Because both drugs were developed to target wild-type EGFR, we hypothesized that current dosing schedules were not optimized for mutant EGFR or to prevent resistance.
To investigate this further, we developed isogenic TKI-sensitive and TKI-resistant pairs of cell lines that mimic the behavior of human tumors.”
What they found was really interesting
In simple terms, they noticed that NSCLC cells grow at different rates, which may possibly explain why some tumours become resistant to EGFR inhibitors faster than others.
What was surprising though, is that EGFR mutant (resistant) cells grew at a slower rate:
“On average, parental cells doubled ~1.22 times faster than T790M-containing resistant cells.”
It isn’t yet clear why this happens though.
In clinical practice, it has been noticed that patients with acquired resistance have re-responded to tyrosine kinase inhibitor (TKI) therapy after a drug holiday. Chmielecki et al., found some evidence as to why this might happen, since they observed that:
“Lysates from parental cells and late-passage PC-9/BR–resistant cells treated with BIBW-2992 showed significantly reduced phosphorylation of EGFR and its downstream targets, extracellular signal–regulated kinase (ERK) and AKT, whereas lysates from resistant cells maintained in the presence of TKI and treated with the same concentrations of drug did not.”
Once the validity of the preclinical findings were established, they looked at evolutionary modelling to design optimal dosing strategies for the use of EGFR inhibitors in NSCLC. They incorporated PK data from clinical trials to ensure the drug doses proposed were feasible. The modelling appeared to be useful:
“This modeling predicted alternative therapeutic strategies that could prolong the clinical benefit of TKIs against EGFR-mutant NSCLCs by delaying the development of resistance.”
It is worth noting the strategy predicted by the model:
“We propose the use of high-dose pulsed once-weekly BIBW-2992 with daily low-dose erlotinib to delay the emergence of T790M-mediated resistance. PC-9 cells treated with this regimen required twice as long to develop resistance and did not show selection for T790M mutations.
In patients, the combination of two EGFR TKIs could lead to overlapping toxicities involving rash and diarrhea. Thus, in a phase IB dose-safety trial, we would recommend a more tolerable strategy, with lower doses of erlotinib still known to be effective against EGFR-mutant tumors (25 or 50 mg daily, orally).”
What’s also fascinating to me is that the overall study findings make sense for consideration when using other TKIs as well, since we know that GIST patients treated with imatinib can re-respond after a period of drug holiday (see Fumagalli et al., (2009). Could different dosing strategies be adopted in some patients at a high risk of developing resistance based on the model approach?
It will be most interesting to see whether clinical trials in lung cancer with EGFR inhibitors evolve along the lines of those suggested by the researchers – that will be the ultimate proof of the pudding that resistance can be influenced in patients with NSCLC – until then, it’s a valuable hypothesis.
References:
![]() Chmielecki, J., Foo, J., Oxnard, G., Hutchinson, K., Ohashi, K., Somwar, R., Wang, L., Amato, K., Arcila, M., Sos, M., Socci, N., Viale, A., de Stanchina, E., Ginsberg, M., Thomas, R., Kris, M., Inoue, A., Ladanyi, M., Miller, V., Michor, F., & Pao, W. (2011). Optimization of Dosing for EGFR-Mutant Non-Small Cell Lung Cancer with Evolutionary Cancer Modeling. Science Translational Medicine, 3 (90), 90-90 DOI: 10.1126/scitranslmed.3002356
Chmielecki, J., Foo, J., Oxnard, G., Hutchinson, K., Ohashi, K., Somwar, R., Wang, L., Amato, K., Arcila, M., Sos, M., Socci, N., Viale, A., de Stanchina, E., Ginsberg, M., Thomas, R., Kris, M., Inoue, A., Ladanyi, M., Miller, V., Michor, F., & Pao, W. (2011). Optimization of Dosing for EGFR-Mutant Non-Small Cell Lung Cancer with Evolutionary Cancer Modeling. Science Translational Medicine, 3 (90), 90-90 DOI: 10.1126/scitranslmed.3002356
E. Fumagalli, P. Coco, C. Morosi, P. Dileo, R. Bertulli, A. Gronchi, & P. G. Casali (2009). Rechallenge with imatinib in GIST patients resistant to second or third line therapy 15th Connective Tissue Oncology Society Meeting, Miami Beach, FL